
Projects
Current Research Projects
Plot by Cecilia González Álvarez
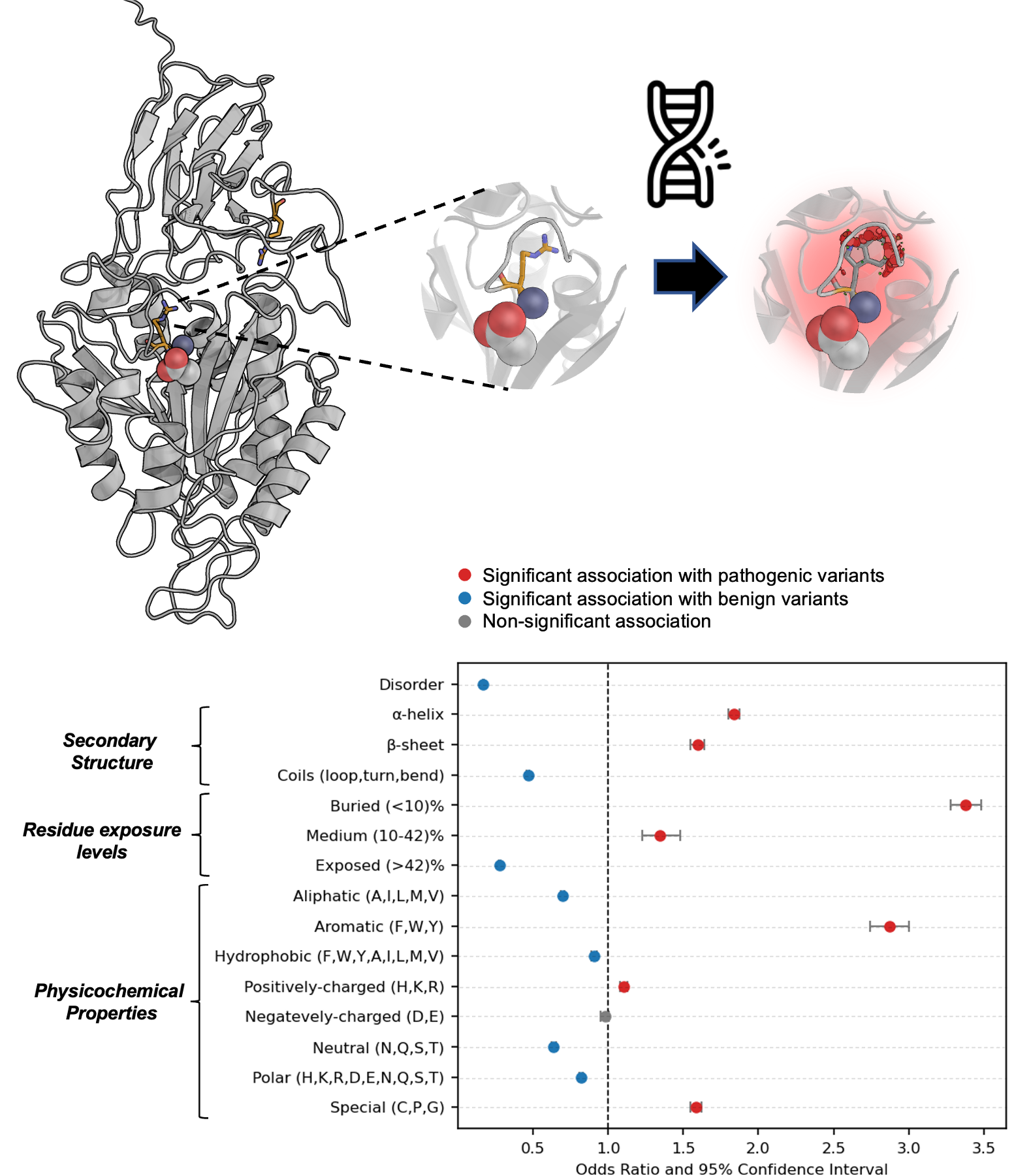
Automating the annotation of protein variants and their relation to disease.
One of my initial aims when starting as a PhD student was the study of protein variants related to disease. There are different mechanisms by which variations can affect a protein’s structure or function, and by precisely identifying genetic variants that are responsible for a patient’s condition, clinicians can design targeted therapeutic strategies that are tailored to each individual’s genetic makeup. This can in turn improve treatment efficacy, avoid adverse side effects, and ultimately lead to better patient outcomes.
During my PhD, I have created a pipeline to annotate protein variants with structure and sequence-based features. Then, I and colleagues studied the structural landscape of pathogenic vs benign variants in different datasets, and identified features characteristic to both classes.
In the future I would really like to turn this project into a tool or service to assists clinicians in the interpretation of protein variants related to disease, with the integration of LLMs.
Manuscript in preparation.
Folding and function of the rice “de novo-ome”
Over the last decade it has been recognized that new genes can emerge de novo from parts of the genome that were previously non-coding, such as intergenic regions, and dramatically enhance protein diversity beyond other well-known mechanisms. Furthermore, it has been shown that de novo genes contribute to the evolution of protein diversity under selective pressure, and de novo gene birth plays a large role in evolutionary innovation.
The process by which de novo genes develop their initial structures, acquire functionality, become established within a population, and persist through multiple speciation events, is still not well understood. To understand how seemingly random sequences can act as starting points for evolutionary innovation, it is necessary to analyze de novo proteins from a structural perspective.
In this study, we compare a group of more than 175 de novo proteins from rice to canonical rice proteins and randomly generated proteins, with the purpose of assessing the degree at which de novo proteins have gene-like characteristics. We find several features that distinguish these groups, and identify potential binding partners and binding topologies against the rice proteome. Collectively, our study furthers our understanding of the origin of protein structure and function, and provides a large-scale comparison of the performance of different AI-based structure prediction tools for cases where no homologous sequences are available.
Manuscript in preparation.



Software Projects
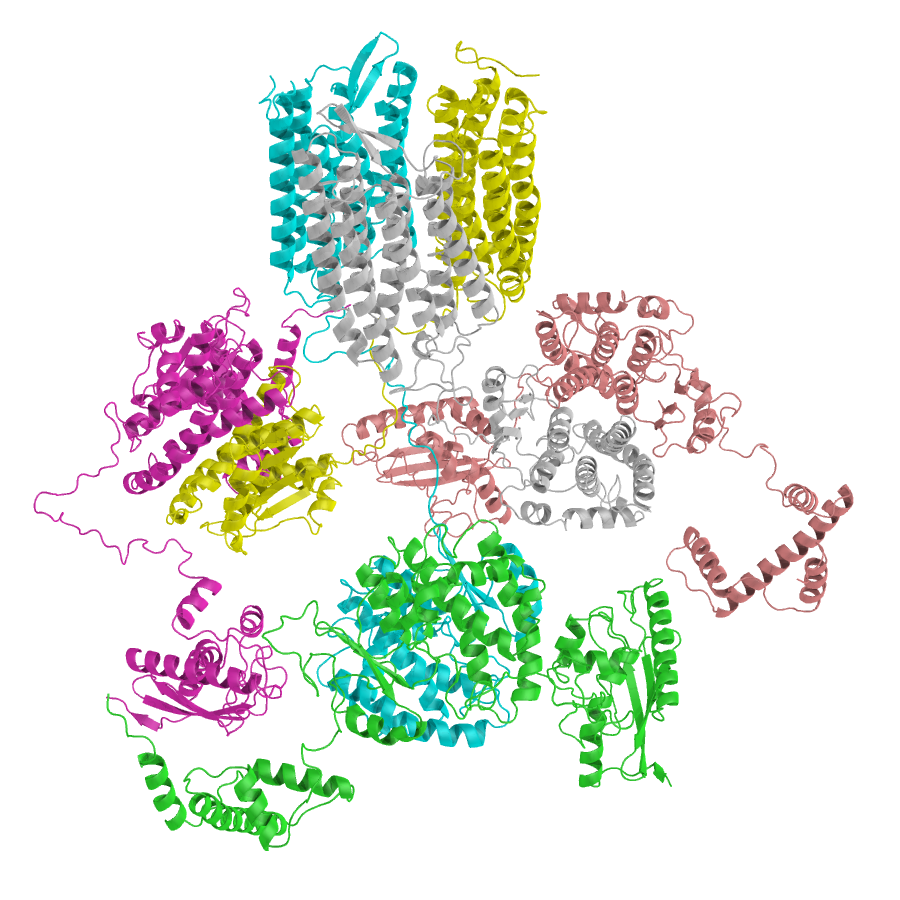
AlphaCRV
Python package that helps to identify correct interactors in a one-against-many AlphaFold-Multimer screen by clustering, ranking, and visualizing conserved binding topologies. It was able to identify the correct binding topologies in 3/3 tested proteome-wide interaction screens.


AlphaFold-Ibex
AlphaFold warpper that allows for easy parallelelization in massive modeling tasks in the Slurm scheduler for HPC clusters. Separates CPU and GPU-bound steps to optimize resources, and provides quality scores and plots to visualize the results.
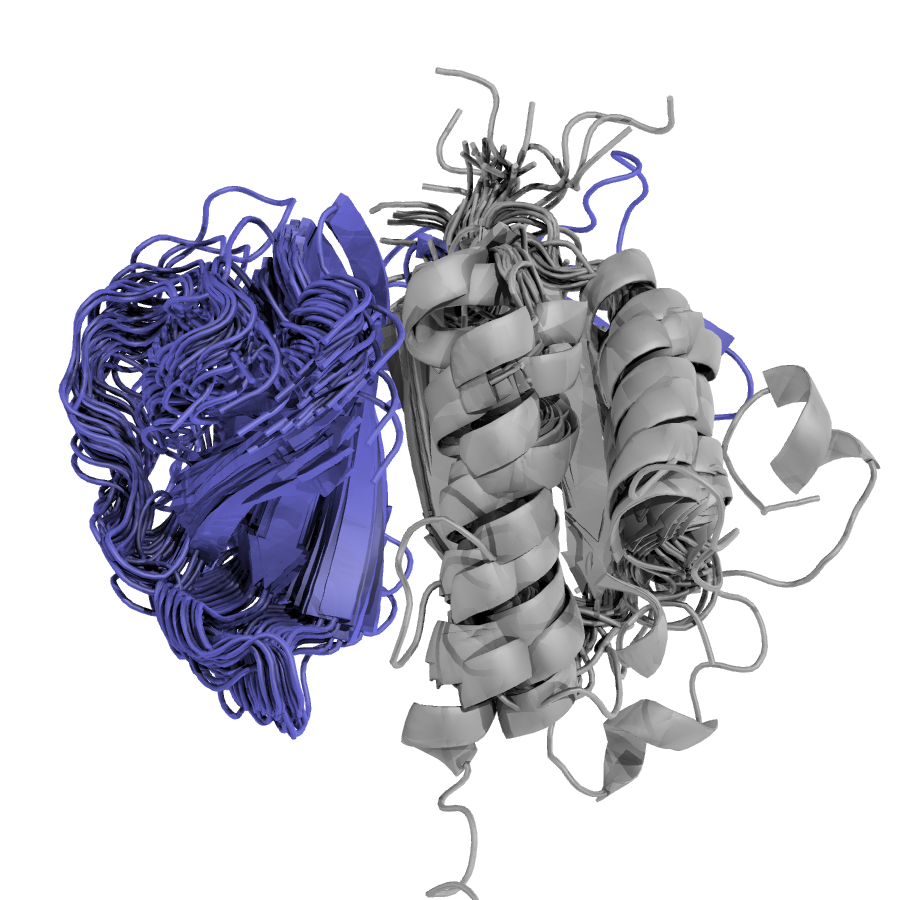
Multiprot
Python-based pipeline to model proteins by connecting two or more structured domains with disordered linkers. Supports single or multiple-chain models and symmetric structures.
CoolerCodonOpt
DNA codon optimization tool using the Python “dnachisel” library. It provides an interface to run codon optimization on one or more protein or DNA sequences.
